Pipeline modules¶
Preprocessing modules¶
Diagnostics¶
These modules can be used to assess the quality of the PAR-CLIP library.
FastQCModule¶
Uses FastQC to create a diagnostic report of various quality aspects of the fastq reads.
Pipeline input: fastq
Pipeline output:
| Parameter | Default value | Description |
|---|---|---|
| kmer_length | 7 | length of the kmers used for detecting overrepresented kmers |
| outdir_name | fastQC | name of the output directory that stores the report |
| extra_flags | [] | list of extra flags passed to the command line tool |
BamPPModule¶
Postprocesses a bam alignment file and creates statistics of mutation rates and mismatch positions. Can clip mutations from the end of the reads.
Pipeline input: bam
Pipeline output: bam
| Parameter | Default value | Description |
|---|---|---|
| remove_n_edge_mut | 0 | clip mutations that are at the n outermost bases in each alignment |
| max_mut_per_read | 1 | discard reads that have more mutations than the given integer |
| min_base_quality | 0 | discard reads that have at least one base with a quality lower than the given integer |
| min_avg_ali_quality | 20 | discard reads that have an average quality lower than the given integer |
| min_mismatch_quality | 20 | discard reads with mismatches of quality lower than the given integer |
| dump_raw_data | False | dump raw data for diagnostic purposes |
| outdir_name | bam_analysis | name of the output directory storing diagnostics and plots |
SoftclipAnalysisModule¶
If the mapper allows soft-clipping this module shows the most frequently clipped sequences.
The output can be used to understand common unclipped contamination. Creates two files
3prime_clipped.txt and 3prime_clipped.txt.
Interpretation of the output:
clipped bases: 2 | clipped sequences: 152
TG 128
CG 7
CT 4
CC 3
TT 3
TC 2
AT 1
CA 1
GG 1
GT 1
The header line contains the number of clipped bases and the number of affected alignments. Below the most commonly clipped sequences are enumerated along with the number of affected alignments in descending order.
Pipeline input: bam
Pipeline output:
| Parameter | Default value | Description |
|---|---|---|
| outdir_name | bam_analysis | name of the output directory storing diagnostics and plots |
TransitionFrequencyModule¶
Plots the frequency of transitions in a bar plot.
Pipeline input: mpileup
Pipeline output:
| Parameter | Default value | Description |
|---|---|---|
| output_prefix | file name prefix of the output file | |
| min_cov | 5 | only consider sites with at least this coverage |
| y_axis_limit | 0 | limit of the y axis |
| remove_tmp_files | True | remove temporary files |
Shows the observed substitution rates for all possible base substitutions. The T->C transitions is displayed as P(C|T).
Example output:
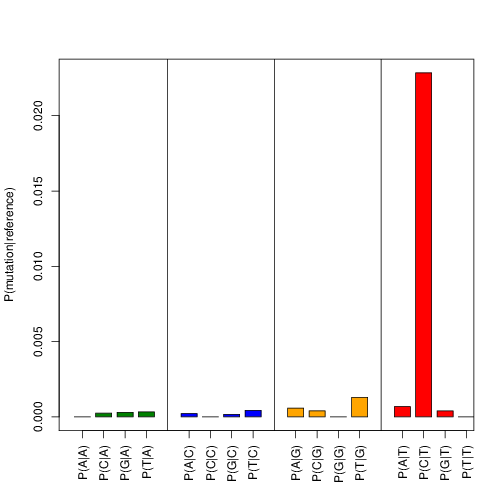
Duplicate removal¶
In order to make meaningful biological conclusions, PCR duplicates should be removed after sequencing. We offer two modules here. A naive approach implemented in DuplicateRemovalModule and two modules using umi_tools, applicable when the library contains unique molecular identifiers (UMIs).
DuplicateRemovalModule¶
A poor man’s approach to duplicate removal: this module removes fastq reads with the same sequence. If the PAR-CLIP library uses UMIs, it is recommended to use the more sophisticated modules based on umi_tools. Please refer to UmiToolsDedupModule and UmiToolsExtractModule.
Pipeline input: fastq
Pipeline output: fastq
UmiToolsExtractModule¶
Together with UmiToolsDedupModule this is the preferred way to deduplicate PAR-CLIP libraries that use UMIs. This module extracts the UMI and appends it to the read name. As such it is recommended to run this module as one of the first steps in a pipeline. After mapping the UmiToolsDedupModule module can be used to remove duplicated reads.
For further information, please also check the documentation of umi_tools [SHS17].
| Parameter | Default value | Description |
|---|---|---|
| extra_flags | [] | list of extra flags passed to the command line tool |
Pipeline input: fastq
Pipeline output: fastq
UmiToolsDedupModule¶
Together with UmiToolsExtractModule this is the preferred way to deduplicate PAR-CLIP libraries that use UMIs. This module deduplicates bam files based on extracted UMIs. This module has to be run after UmiToolsExtractModule.
For further information, please also check the documentation of umi_tools [SHS17].
Pipeline input: bam
Pipeline output: bam
| Parameter | Default value | Description |
|---|---|---|
| extra_flags | [] | list of extra flags passed to the command line tool |
Adapter clipping¶
Adapter clipping is an important step in PAR-CLIP libraries: in case of very small inserts, the beginning of the 3’ adapter is present in the reads. SkewerAdapterClippingModule is the module of choice for removing these adapters.
ClippyAdapterClippingModule clips adapters with less sensitity, but also detects partial 5’ adapters. It can be useful when dealing with libraries that were generated from very low amounts of RNA.
SkewerAdapterClippingModule¶
This module uses skewer [JLDZ14] to trim the 3’ adapter from the PAR-CLIP reads. Additional arguments can be directly passed to skewer’s command line call. Please consult skewer’s documentation for a detailed description of all available options.
Pipeline input: fastq
Pipeline output: fastq
| Parameter | Default value | Description |
|---|---|---|
| extra_args | [] | list of extra flags passed to the command line tool |
ClippyAdapterClippingModule¶
This module removes longer traces of adapters by looking for perfect matches of the adapter ends.
It also removes random barcodes and adapters. If you are using UmiToolsExtractModule to
extract the UMIs don’t forget to set clipped_5prime_bc to True.
Pipeline input: fastq
Pipeline output: fastq
| Parameter | Default value | Description |
|---|---|---|
| clip_len | 10 | minimum base pairs required to be detected as adapter sequence |
| clipped_5prime_bc | False | UMIs already removed from the 5’ end |
Mapping¶
With Bowtie and STAR we offer two very different alignment strategies. STAR can map spliced reads and use softclipping to remove contaminants automatically. Soft clipping however prevents from mapping transitions at either end of the alignment. Mappers feed unmapped reads back in the pipeline and thus can be chained.
STARMapModule¶
STAR [DDS+13] is a general purpose RNA-seq data mapper. Unmapped reads are returned to the pipeline in fastq format. For detailed configuration options, please also refer to STAR’s user manual.
Pipeline input: fastq
Pipeline output: bam, fastq
| Parameter | Default value | Description |
|---|---|---|
| genome_index | path to the directory containing the STAR genome index | |
| n_mismatch | 1 | maximum number of allowed mismatches |
| n_multimap | 1 | maximum number of mapping positions. Maps uniquely by default |
| allow_soft_clipping | True | enable softclipping |
| outdir_name | star_out | name of the output directory |
| extra_flags | [] | additional commandline options passed to STAR |
BowtieMapModule¶
Bowtie [Lan10] is a genomic aligner and as such cannot map spliced reads. Unmapped reads are re-queued in the pipeline.
Pipeline input: fastq
Pipeline output: bam, fastq
| Parameter | Default value | Description |
|---|---|---|
| genome_index | prefix of bowtie’s genome index | |
| n_mismatch | 1 | maximum number of allowed mismatches |
| n_multimap | 1 | maximum number of mapping positions. Maps uniquely by default |
| extra_flags | [] | additional commandline options passed to bowtie |
Binding site prediction¶
We offer two different strategies for predicing binding sites. BSFinderModule does not require mock information and therefore cannot distinguish background binding from factor specific binding events. MockinbirdModule is the recommended binding site predictor. It requires parameters trained on a mock experiment.
BSFinderModule¶
BSFinder calculates p-values by learning a statistical model on non-specific conversion events. This model cannot distinguish background binding from factor specific binding events. The prediction algorithm was presented in [Tor15].
Pipeline input: mpileup
Pipeline output: table
| Parameter | Default value | Description |
|---|---|---|
| pval_threshold | 0.005 | only sites with a p-value smaller than this are reported |
| min_cov | 2 | minimum coverage of reported binding sites |
NaiveBSFinderModule¶
The naive binding site finder predicts all sites that pass a specified minimum threshold of conversion events.
Pipeline input: mpileup
Pipeline output: table
| Parameter | Default value | Description |
|---|---|---|
| min_transitions | 2 | only sites with at least this many conversion events are reported |
MockinbirdModule¶
Mockinbird is the core module that predicts binding sites by harnessing information from a mock experiments. Its input files are generated by the modules in the Mockinbird related modules section.
Pipeline input: trtable, mock_model
Pipeline output: table
| Parameter | Default value | Description |
|---|---|---|
| plot_dir | mockinbird_plots | directory for writing out diagnostic plots |
| max_k_mock | 10 | sites with more specific conversions than max_k_mock are
discarded |
| extra_args | [] | additional arguments directly passed to the called script |
Miscellaneous¶
A collection of miscellaneous helper modules.
SortIndexModule¶
The SortIndexModule sorts and indexes a bam files using samtools. This is generally required before generating a pileup file.
Pipeline input: bam
Pipeline output: bam
PileupModule¶
Uses samtools to create a pileup file. Pileup report coverage and transitions per genomic base and are the input of our predictors. Please be aware that the bam file has to be sorted. If in doubt, queue after the SortIndexModule.
Pipeline input: bam
Pipeline output: mpileup
NormalizationModule¶
The normalization module calculates an occupancy by dividing the number of observed transitions by the coverage of a reference experiment. The appropriate reference experiment should reflect the pool of RNA the factor sees when choosing where to bind. Depending on the binding properties of the protein of interest, an RNA-seq experiment under PAR-CLIP conditions, PAR-CLIP of the RNA polymerase or protocols to capture transient binding such as 4SU-seq may be appropriate.
Additionally, SNPs are removed by detecting elevated conversion rates in the normalization pileup file.
Pipeline input: table
Pipeline output: table
| Parameter | Default value | Description |
|---|---|---|
| mut_snp_ratio | 0.75 | ratio of conversations to coverage in the normalization pileup for a site being detected as SNP |
QuantileCapModule¶
Caps the occupancy value at a given quantile. This module can help removing the influence of outliers on downstream analyses, such as the gene plot.
Pipeline input: table
Pipeline output: table
| Parameter | Default value | Description |
|---|---|---|
| max_quantile | 0.95 | all occupancy values are capped to the value of this quantile |
Table2FastaModule¶
Converts a binding site table file to fasta by extracting the genomic sequence around the binding site.
Pipeline input: table
Pipeline output: fasta
| Parameter | Default value | Description |
|---|---|---|
| genome_fasta | path to the genome fasta file |
Postprocessing modules¶
Plots¶
CenterPlotBSModule¶
This module plots a bootstrapped metagene plot fixed at the start and end of each annotations.
Pipeline input: table
Pipeline output:
| Parameter | Default value | Description |
|---|---|---|
| gff_file | gff file of annotations used for the metagene plot | |
| output_prefix | file name prefix for the output files | |
| labelCenterA | label for the metagene start position | |
| labelCenterB | label for the metagene end position | |
| labelBody | label for the metagene body | |
| downstream_bp | 1000 | number of base pairs shown downstream of start |
| upstream_bp | 1000 | number of base pairs shown upstream of the end |
| gene_bp | 750 | number of base pairs inside the annotation, i.e. downstream of the start and upstream of the end |
| min_trscr_size_bp | 1500 | filter out all transcript shorter than this size |
| max_trscr_size_bp | 100000 | filter out all trascript longer than this size |
| smoothing_window | 20 | size of the window used for smoothing the profile (in bp) |
| remove_tmp_files | True | clean up temporary files |
| bootstrap_iter | 2500 | number of bootstrap iterations |
| n_processes | 4 | number of parallel processes |
Example plot:
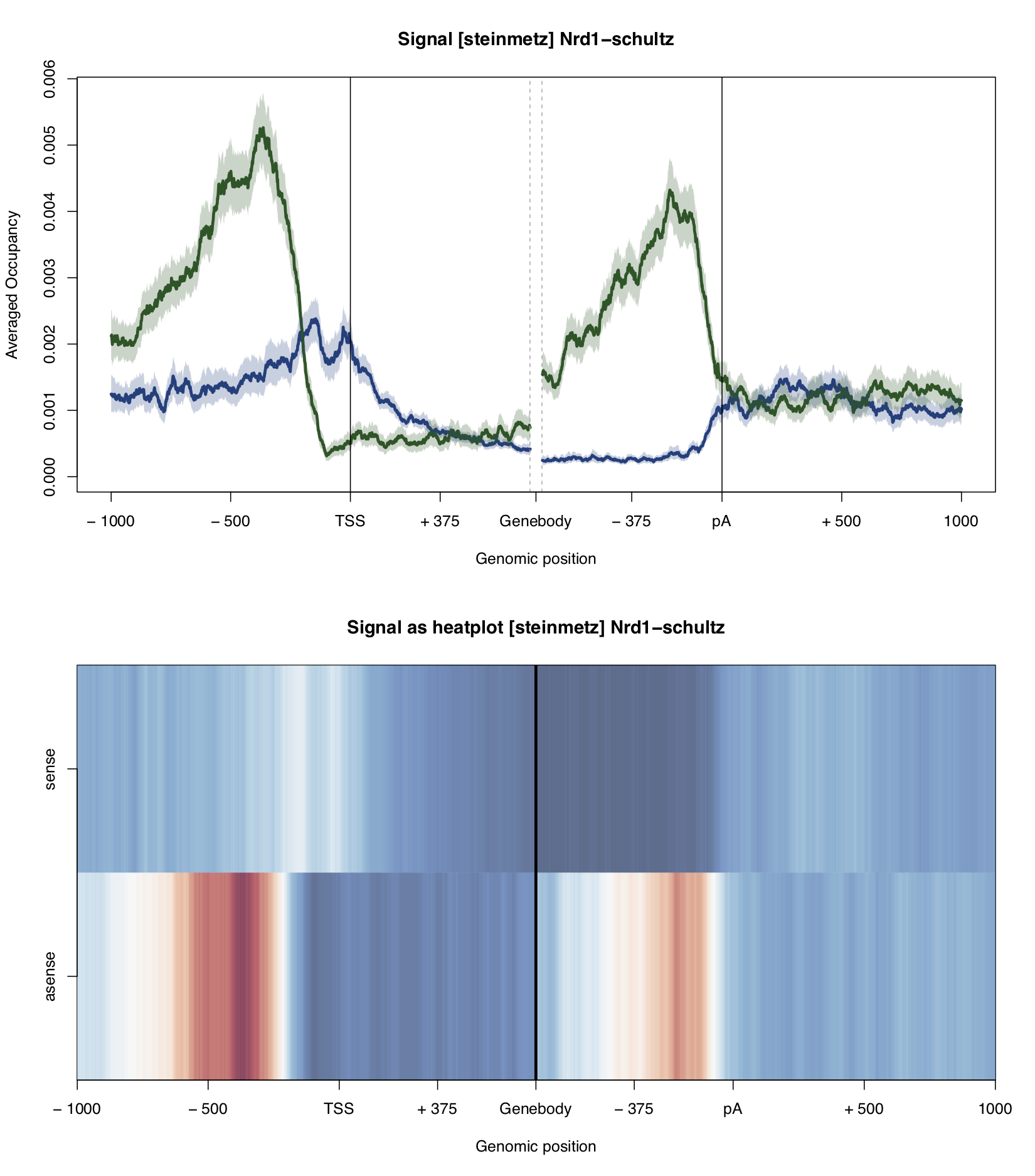
A plot of the PAR-CLIP occupancy around the start and end sites of genomic annoations. The occupancy profile of the sense strand is depicted in blue, occupancy on the anti-sense strand is shown in green. The profiles are smoothed with a running mean. 95%-confidence intervals are calculated by bootstrap sampling the annotations.
The bottom of the plot shows the occupancy profiles in heatmap representation.
KmerPerPositionModule¶
Plots the kmer occurence frequencies around binding sites.
Pipeline input: table
Pipeline output:
| Parameter | Default value | Description |
|---|---|---|
| genome_fasta | path to genome in fasta format (requires index) | |
| output_prefix | file name prefix for the output files | |
| kmer_k | 3 | length of kmers |
| first_index | 0 | index of the first site plotted |
| last_index | 1500 | index of the last site plotted |
| width | 50 | number of base pairs around the binding site considered |
| sort_key | occupancy | sort column of the table; valid values are occupancy, transitions, coverage and score. |
| gff_exclude_path | ‘’ | sites that overlap one of the annotations in this file are dropped |
| gff_padding | 20 | annotations are extended by this amount of base pairs around start and end |
| remove_tmp_files | True | clean up temporary files |
HeatmapPlotModule¶
Plot a heatmap of all transcripts matching given length criteria as a heat map. Using binning over transcripts and transcript lenth.
Pipeline input: table
Pipeline output:
| Parameter | Default value | Description |
|---|---|---|
| gff_file | gff file of annotations used for the metagene plot | |
| output_prefix | file name prefix for the output files | |
| downstream_bp | 4000 | number of base pairs shown downstream of start |
| upstream_bp | 1000 | number of base pairs shown upstream of the end |
| min_trscr_size_bp | 0 | filter out all transcript shorter than this size |
| max_trscr_size_bp | 5000 | filter out all transcripts longer than this size |
| x_bins | 500 | number of bins in x direction (transcript length) |
| y_bins | 500 | number of bins in y direction (grouping transcripts) |
| x_pixels | 500 | pixel in x directions |
| y_pixels | 500 | pixels in y direction |
| remove_tmp_files | True | clean up temporary files |
HeatmapSmallPlotModule¶
Plot a heatmap of all transcripts matching given length criteria as a heat map. Using binning over transcripts and transcript lenth. Similar to HeatmapPlotModule, but more consise plot.
Pipeline input: table
Pipeline output:
| Parameter | Default value | Description |
|---|---|---|
| gff_file | gff file of annotations used for the metagene plot | |
| output_prefix | file name prefix for the output files | |
| downstream_bp | 500 | number of base pairs shown downstream of start |
| upstream_bp | 1000 | number of base pairs shown upstream of the end |
| min_trscr_size_bp | 0 | filter out all transcript shorter than this size |
| max_trscr_size_bp | 5000 | filter out all transcripts longer than this size |
| x_bins | 500 | number of bins in x direction (transcript length) |
| y_bins | 500 | number of bins in y direction (grouping transcripts) |
| x_pixels | 500 | pixel in x directions |
| y_pixels | 500 | pixels in y direction |
| remove_tmp_files | True | clean up temporary files |
Motif detection¶
XXmotifModule¶
Runs XXmotif, a tool for de-novo detection of overrepresented motifs.
Pipeline input: table
Pipeline output:
| Parameter | Default value | Description |
|---|---|---|
| genome_fasta | path to genome in fasta format (requires index) | |
| output_prefix | file name prefix for the output files | |
| negative_set_gff | path to a gff file used for sampling the negative set | |
| n_negative seqs | 20000 | number of negative sequences sampled |
| first_index | 0 | index of the first site plotted |
| last_index | 1500 | index of the last site plotted |
| width | 12 | number of base pairs around the binding site considered |
| sort_key | occupancy | sort column of the table; valid values are occupancy, transitions, coverage and score. |
| gff_exclude_path | ‘’ | sites that overlap one of the annotations in this file are dropped |
| gff_padding | 20 | annotations are extended by this amount of base pairs around start and end |
| remove_tmp_files | True | clean up temporary files |
Miscellaneous¶
GffFilterModule¶
Filter sites that overlap with a given gff annotation.
Can be used to filter sites in highly abundant transcripts such as tRNA or rRNA.
Pass the name of the gff features you want to exclude to the features option.
Pipeline input: table
Pipeline output: table
| Parameter | Default value | Description |
|---|---|---|
| filter_gff | path to gff file used for filtering | |
| file_postfix | fil | postfix appended to the table name |
| padding_bp | 20 | annotations are extended by this amount of base pairs |
| features | [] | list of features that are filtered. Excludes all by default. |
Writing your own modules¶
A straightforward way to implement your own pipeline module is by subclassing
CmdPipelineModule. Here we use the SkewerAdapterClippingModule as an example.
from mockinbird.utils import pipeline as pl
class SkewerAdapterClippingModule(pl.CmdPipelineModule):
Adding module arguments¶
If your module accepts arguments, you have to modify the constructor __init__(self, pipeline):
def __init__(self, pipeline):
cfg_fmt = [
('extra_args', cv.Annot(list, default=[])),
]
super().__init__(pipeline, cfg_req=cfg_fmt)
Each argument is a tuple consisting of a name, here extra_args and an annotation object
Annot. Upon construction the annotation takes following keyword arguments:
- type:
- a callable that represents the base type of the value, here a
list - default:
- the default value if the option was not provided by the user. The default value
Nonemakes setting the value in the configuration file mandatory. - converter:
- a callable that validates and converts the value the user set in the config file.
Can raise a
ValueError, if the user entered an invalid value.
By calling super().__init__() the list of arguments cfg_fmt is passed to the parent
constructor.
Following configurations are now valid in an configuration file:
[...]
- SkewerAdapterClippingModule:
extra_args:
- -k 12
- -d 0.01
[...]
- SkewerAdapterClippingModule:
extra_args: []
which is equivalent to falling back to the default argument
[...]
- SkewerAdapterClippingModule
We define a variety of validators that can readily be used. Please refer to Configuration for details.
Preparing the module¶
prepare(self, cfg) is the heart of the module and defines the commands that are executed when
the module runs. It also registers the outputs and thus makes new files visible to downstream
modules.
cfg is a dict of all configuration options set by the user. Configuration of all options
requested as described in Adding module arguments can be accessed by their names.
The first action in the prepare method has to be the call to the parent’s prepare method:
def prepare(self, cfg):
super().prepare(cfg)
Information from previously run modules and global configuration options can be obtained through
a reference to Pipeline, which can be accessed through the private attribute
self._pipeline.
def prepare(self, cfg):
super().prepare(cfg)
pipeline = self._pipeline
general_cfg = pipeline.get_config('general')
read_cfg = pipeline.get_config('reads')
output_dir = general_cfg['output_dir']
prefix = general_cfg['prefix']
Here we access the global configuration sections general and reads to obtain the path to
the output directory and the file name prefix.
Pipeline.get_curfile() is used to obtain the path to files that are queued in
the pipeline.
The pipeline stores the most recent file path of each format.
def prepare(self, cfg):
[...]
fastq_file = pipeline.get_curfile(fmt='fastq')
Here we obtain the path to the most recently queued fastq file.
Now we have all information to construct the command line call for running skewer.
The output path is stored in adapter_clipped_file, a file path relative to the
output directory passed by the command line script.
def prepare(self, cfg):
[...]
adapter_clipped_file = os.path.join(output_dir, prefix + '_skewer.clipped')
cmd = [
'skewer',
fastq_file,
'-x %s' % general_cfg['adapter3prime'],
'-m tail',
'--min %s' % read_cfg['min_len'],
'--quiet',
'--stdout',
'> %r' % adapter_clipped_file,
]
if cfg['extra_args']:
cmd.extend(cfg['extra_args'])
self._cmds.append(cmd)
Having collected the command as a list of arguments in cmd, we append all user defined
arguments that are conveniently stored in cfg and queue the command by appending it to the
private self._cmds variable.
self._intermed_files.append(adapter_clipped_file)
pipeline.upd_curfile(fmt='fastq', filepath=adapter_clipped_file)
As a final step we register adapter_clipped_file as an intermediate file, meaning that it
can be deleted provided this module is not the last in the pipeline.
Pipeline.upd_curfile() registers the file as new fastq file and thus
makes it visible to following modules.
Note: At time prepare() is called, files to the passed paths may or may not exist. Only queue new commands by appending to self._cmds, never try to execute commands.
The full module¶
Putting everything together, the code of the SkewerAdapterClippingModule looks like this:
import os
from mockinbird.utils import pipeline as pl
from mockinbird.utils import config_validation as cv
class SkewerAdapterClippingModule(pl.CmdPipelineModule):
def __init__(self, pipeline):
cfg_fmt = [
('extra_args', cv.Annot(list, default=[])),
]
super().__init__(pipeline, cfg_req=cfg_fmt)
def prepare(self, cfg):
super().prepare(cfg)
pipeline = self._pipeline
general_cfg = pipeline.get_config('general')
read_cfg = pipeline.get_config('reads')
output_dir = general_cfg['output_dir']
prefix = general_cfg['prefix']
fastq_file = pipeline.get_curfile(fmt='fastq')
adapter_clipped_file = os.path.join(output_dir, prefix + '_skewer.clipped')
cmd = [
'skewer',
fastq_file,
'-x %s' % general_cfg['adapter3prime'],
'-m tail',
'--min %s' % read_cfg['min_len'],
'--quiet',
'--stdout',
'> %r' % adapter_clipped_file,
]
if cfg['extra_args']:
cmd.extend(cfg['extra_args'])
self._cmds.append(cmd)
self._intermed_files.append(adapter_clipped_file)
pipeline.upd_curfile(fmt='fastq', filepath=adapter_clipped_file)
The parent class CmdPipelineModule and the Pipeline take care of all required
steps such as config parsing and validation, executing commands and cleaning up files.
Custom modules in config files¶
Having created your own module, you have to make the class importable in python.
The cleanest way to achieve that is to make your module installable by writing a
setup.py. For more information please refer to available documentation, e.g.
here.
In the config file you have to refer to the module with its full import path, e.g.:
- mypackage.mymodule.SkewerAdapterClippingModule:
extra_args:
- -k 2